研究者名:佐谷 秀行
プロジェクト概要
増殖能を持つ正常細胞は、複製・分裂の途上において遺伝子や染色体の異常が生じると、一時的に細胞周期を停止しその異常を修正する(あるいは異常細胞を除去する)能力があり、組織内に異常細胞が出現することを防いでいると考えられている。この機構をチェックポイントと呼ぶ。細胞周期の各フェーズに設定されたチェックポイントは、様々な細胞内分子が連係することにより制御されており、それらの分子自身あるいは連係に異常が生じることにより、多発性の遺伝子変異や染色体の不均等分配など「ゲノム不安定化」が発生し、細胞の腫瘍化ならびに悪性化が生じてくると考えられている。
細胞分裂は染色体と細胞質内の要素を正確に二分する重要なステップである。したがって細胞分裂をひかえたG2期からM期進行の調節機構破綻は細胞の形質にドラスティックな変化を引き起こし、ゲノムの不安定化を誘発する。G2期からM期の進行は(1)タンパク分解、(2)タンパクリン酸化という2つの生化学的イヴェントによって制御されていることが近年の研究により明らかにされつつある。主として、前者においてはanaphase promoting complex (APC)と呼ばれるユビキチンリガーゼ、後者ではmitotic kinasesと呼ばれる一群のリン酸化酵素が重要な役割を演じていることが酵母などを用いた研究によって明らかにされている。私達はこれらAPC及びmitotic kinasesの機能ならびに活性の分子制御について検討し、これらの制御破綻が細胞の悪性化に働く可能性について研究を行なっている。
関連論文
1) Nishiyama et al.: FEBS Letters (1999) 459, 158-165
2) Hirota et al.: J Cell Biol (2000) 149: 1073-1086
3) Tsuiki et al.: Oncogene (2001) 20: 420-429
4) Sudo et al.: EMBO J (2001) 20: 6499-6508
研究者名:佐谷 秀行
プロジェクト概要
CD44はヒアルロン酸を始めとする細胞外マトリックスと結合する接着分子であり、1.リンパ球ホーミング、2.リンパ球活性化、3.細胞-細胞間接着及び細胞-基質間接着、4.細胞運動、5.癌細胞増殖・転移などに深く関与している。そしてその機能は発現量だけでなく、alternative splicingによるバリアントアイソフォームの発現1)2)や、糖鎖付加やリン酸化といったいわゆる翻訳後修飾によっても制御される。我々は中でもCD44細胞外ドメインのproteolytic cleavage(切断)による機能調節に注目して研究を進めている。
リンパ球やある種の癌細胞では培養上清中に可溶型CD44(soluble CD44)と呼ばれる細胞内領域及び膜貫通領域を欠いたCD44が存在することが知られている。このsoluble CD44は実際に癌患者の血清中にも検出され、胃癌や乳癌患者では血清soluble CD44値と病期との間に相関を認めることが報告されている。実験的にも高転移性のメラノーマ細胞株では培養上清中に大量のsoluble CD44が検出されるが、同じ細胞由来の低転移株では殆ど検出されないことから3)、soluble CD44の増加は癌の進展の指標となりうるのではないかと予想されていた。近年我々は、このsoluble CD44はCD44が細胞外領域にて膜型メタロプロテアーゼによってcleavageされた後のN端フラグメントであることを発見した。さらにこのCD44 cleavageをメタロプロテアーゼ阻害剤によって抑制すると、CD44のリガンドであるヒアルロン酸上での癌細胞運動能が抑制されることから、CD44 cleavageという現象そのものが癌細胞運動に大変重要であることが分かった4)。
更に我々はCD44の細胞外ドメインのcleavageはL-selectinやproHB-EGFなどの膜蛋白と同様にTPA及びカルシウム・イオノフォア処理によって誘発されることを見い出した5)。TPAによるCD44 cleavageはPKC活性化を介するが、カルシウム・イオノフォアはPKCを介さないことが明らかになり、独立したシグナル経路でCD44 cleavage及び細胞運動を制御していることが分かった。また、TPAは低分子GTP結合蛋白質Racを介してruffling形成を促進し、同時にRhoを不活化することによってストレスファイバーを消失させることが知られているが、興味深いことにCD44はこの際生じたrufflingに集積しており(図1)、TPA依存性のCD44 cleavageにはRacの活性化が関与していることが示唆された。
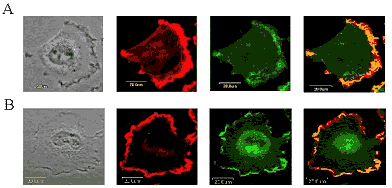
図1 TPA刺激によるアクチンフィラメントの再構成、及びそれに伴うCD44の再局在化
U251MG細胞をTPA刺激30分後に固定(B)し、アクチンをRhodamine-Phalloidinにて、CD44を間接蛍光抗体法(FITC)にてそれぞれ染色。非刺激下(A)と比較してストレスファイバーの消失及び細胞膜全周性にruffling形成を認め、ruffling部に一致したCD44の再局在化を認める。
Okamoto I, et al: J Biol Chem (1999) 36: 25525-25534より改変
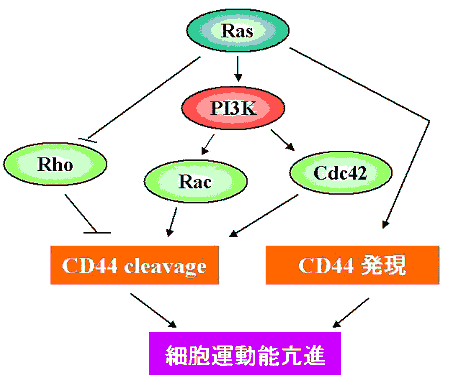
図2 Ras活性化によるCD44を介した細胞運動能亢進の機序
また私達はCD44がその細胞外領域において切断を受けた後、連続的に細胞膜貫通領域でも切断が生じることを見い出した(7)。この切断によって細胞膜より遊離したCD44の細胞内領域(CD44ICD)は核内へ移行し、ルシフェラーゼアッセイにおいてTREのプロモーター配列を介して転写活性を上昇させた。さらに、CD44ICDはTREに直接結合する転写因子ではなくcoactivatorであるCBP/p300を介して転写を制御しており、adenovirus E1Aによってその転写活性は阻害された。また、CD44ICDを導入することにより、内因性のCD44自身の転写が上昇することを見出し、 CD44の切断が新たなCD44分子の合成を促進することが示唆された。これらの結果より、CD44分子は機械的な接着のみならずシグナルを伝達する分子として働いていることが明らかになった。癌細胞におけるCD44の発現の意義や機能については未だ充分に解明されていない点が多いが、CD44の切断機構およびそれを介する癌細胞浸潤の分子機構をさらに明らかにしていく事が今後の癌転移治療の開発にも有用であると考えている。
関連論文
1) Sasaki J, et al: Int J Oncol (1998) 12: 525-533
2) Okamoto I, et al: J Natl Cancer Inst (1998) 90: 307-315
3) Goebeler M, et al: J Cell Sci (1996) 109: 1957-1964
4) Okamoto I, et al: Oncogene (1999) 18:1435-1446
5) Okamoto I, et al: J Biol Chem (1999) 36: 25525-25534
6) Kawano Y, et al: J Biol Chem (2000) 38: 29628-29635
7) Okamoto I, et al: J Cell Biol (2001) 155: 755-762